
We have introduced Biomedical and Line DoD Technology Readiness Levels (TRLs) and discussed how a technology may develop through each stage of the TRL scale for biomedical products such as medical devices. In this article, we will focus on how biomedical TRLs have been adapted for use with medical countermeasures (MCMs) and the unique requirements for development of MCM products, such as drugs and biologics.
Medical Countermeasures (MCMs)
Medical countermeasures (MCMs) are developed to prepare, protect against, and detect natural and man-made chemical, biological, radiological, and nuclear (CBRN) public health threats (e.g., drugs, vaccines, diagnostics). The processes of developing MCM products or Product Development Tools (PDTs; such as assays, models, and reagents) are two distinct but closely related endeavors. In order to successfully develop an MCM, appropriate effort and funding must be directed towards the maturation of the analytical tools and animal models that will be used during MCM development. Similarly, the necessary in vitro and in vivo assays and models cannot be fully refined and verified without a candidate product to provide a substrate for evaluation. These development efforts must proceed in parallel. For more information, visit MedicalCountermeasures.gov.
The Animal Rule
Commercial developers of MCMs face the difficult question of how to obtain regulatory approval for new products. There are important ethical concerns about conducting human clinical trials with bioterrorism agents and additional regulatory complication generates increased commercial risk. The FDA “Animal Rule” allows sponsors to gain regulatory approval for new MCMs on the basis of animal studies that model the disease in human beings. The rule stipulates that animal studies can be used to establish effectiveness for products where:
- The mechanisms of toxicity of the product are well understood;
- The effect is established in more than one species of animal expected to be predictive for humans (or one well characterized animal model); and
- The workings of the drug are sufficiently well understood to allow for the selection of an effective dose in humans.
MCM TRLs
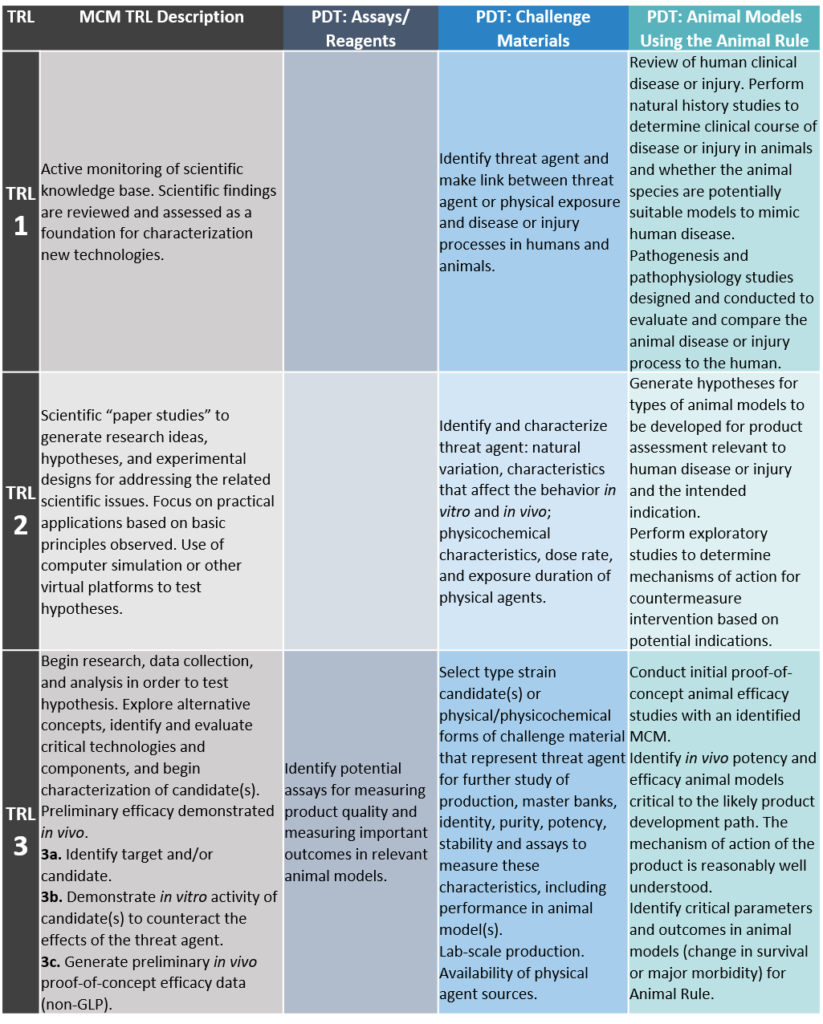
TRLs have been designed and defined for evaluating the maturity of MCM products and PDTs by the Public Health Emergency Medical Countermeasures Enterprise. Agency-specific listings of MCM TRLs by product type can also be found in some broad agency announcements (BAAs) from the Biomedical Advanced Research and Development Authority (BARDA BAA-18-100-SOL-00003) and Defense Threat Reduction Agency (DTRA). A MCM product should be rated at a particular level (or sub-level) only after the sponsor has completed all activities listed in that level. The TRLs for MCM products are detailed below:
TRL 1: Review of Scientific Knowledge Base
Active monitoring of scientific knowledge base. Scientific findings are reviewed as a foundation for characterizing new technologies.

TRL 2: Development of Hypotheses and Experimental Designs
Scientific “paper studies” to generate research ideas, hypotheses, and experimental designs for addressing the related scientific issues. Focus on practical applications based on basic principles observed. Use of computer simulation or other virtual platforms to test hypotheses.

TRL 3: Target/Candidate Identification and Characterization of Preliminary Candidate(s)
Begin research, data collection, and analysis in order to test hypothesis. Explore alternative concepts, identify and evaluate critical technologies and components, and begin characterization of candidate(s). Preliminary efficacy demonstrated in vivo.
3a. Identify target and/or candidate.
3b. Demonstrate in vitro activity of candidate(s) to counteract the effects of the threat agent.
3c. Generate preliminary in vivo proof-of-concept efficacy data (non-GLP [Good Laboratory Practice]).

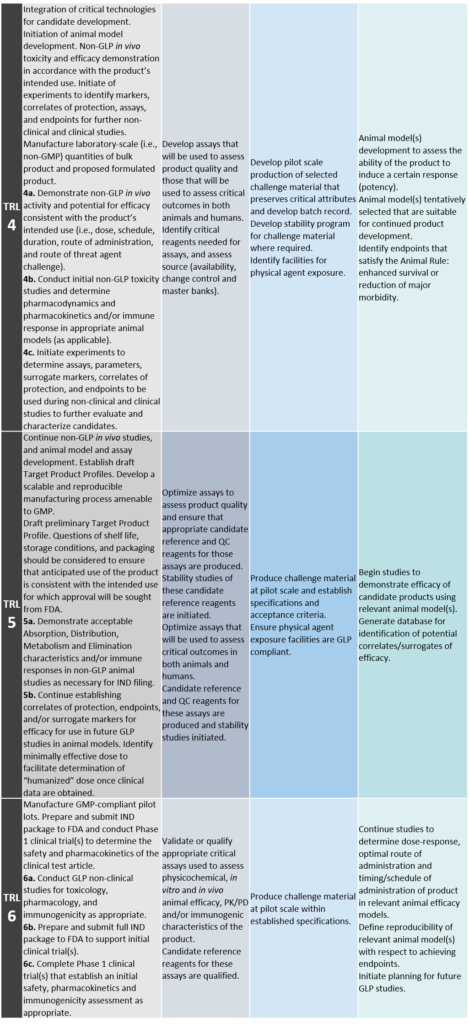
TRL 4: Candidate Optimization and Non-GLP In Vivo Demonstration of Activity and Efficacy
Integration of critical technologies for candidate development. Initiation of animal model development. Non-GLP in vivo toxicity and efficacy demonstration in accordance with the product’s intended use. Initiation of experiments to identify markers, correlates of protection, assays, and endpoints for further non-clinical and clinical studies.
Manufacturing: Manufacture laboratory-scale (i.e., non-GMP [Good Manufacturing Practice]) quantities of bulk product and proposed formulated product.
4a. Demonstrate non-GLP in vivo activity and potential for efficacy consistent with the product’s intended use (i.e., dose, schedule, duration, route of administration, and route of threat agent challenge).
4b. Conduct initial non-GLP toxicity studies and determine pharmacodynamics and pharmacokinetics and/or immune response in appropriate animal models (as applicable).
4c. Initiate experiments to determine assays, parameters, surrogate markers, correlates of protection, and endpoints to be used during non-clinical and clinical studies to further evaluate and characterize candidates.

TRL 5: Advanced Characterization of Candidate and Initiation of GMP Process Development
Continue non-GLP in vivo studies, and animal model and assay development. Establish draft Target Product Profiles. Develop a scalable and reproducible manufacturing process amenable to GMP.
Target Product Profile: Draft preliminary Target Product Profile. Questions of shelf life, storage conditions, and packaging should be considered to ensure that anticipated use of the product is consistent with the intended use for which approval will be sought from FDA.
5a. Demonstrate acceptable Absorption, Distribution, Metabolism and Elimination characteristics and/or immune responses in non-GLP animal studies as necessary for IND filing.
5b. Continue establishing correlates of protection, endpoints, and/or surrogate markers for efficacy for use in future GLP studies in animal models. Identify minimally effective dose to facilitate determination of “humanized” dose once clinical data are obtained.

TRL 6: GMP Pilot Lot Production, IND Submission, and Phase 1 Clinical Trial(s)
Manufacture GMP-compliant pilot lots. Prepare and submit Investigational New Drug (IND) package to FDA and conduct Phase 1 clinical trial(s) to determine the safety and pharmacokinetics of the clinical test article.
6a. Conduct GLP non-clinical studies for toxicology, pharmacology, and immunogenicity as appropriate.
6b. Prepare and submit full IND package to FDA to support initial clinical trial(s).
6c. Complete Phase 1 clinical trial(s) that establish an initial safety, pharmacokinetics and immunogenicity assessment as appropriate.

- Submit protocols for Phase 2 trials to FDA for review.
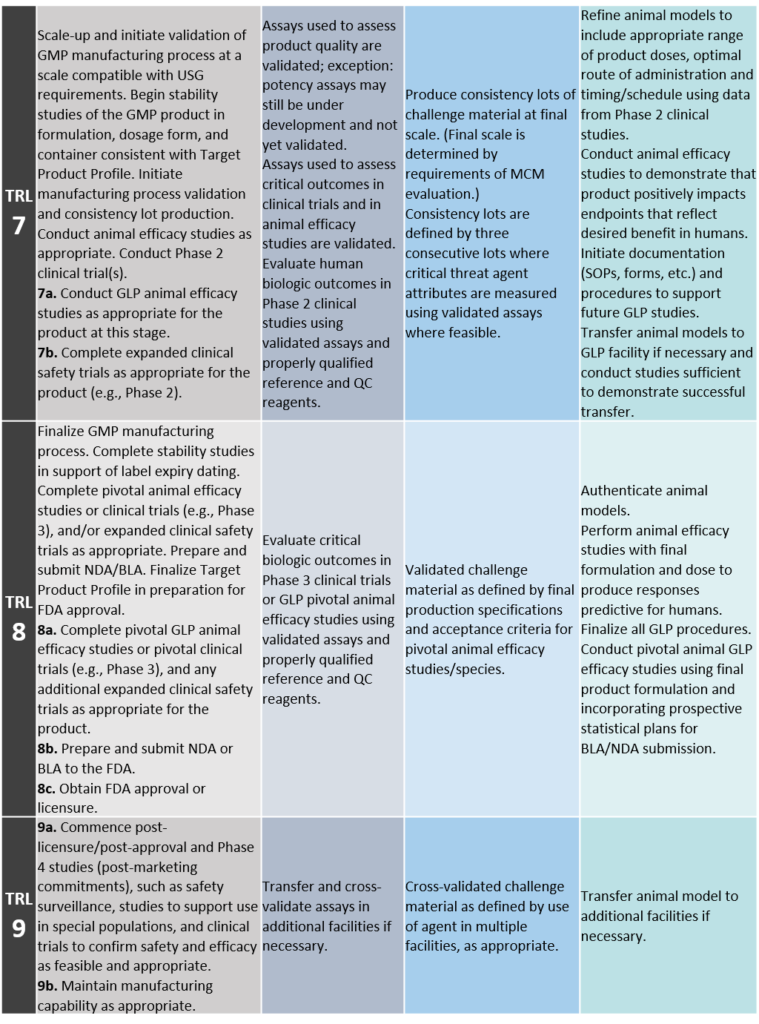
TRL 7: Scale-up, Initiation of GMP Process Validation, and Phase 2 Clinical Trial(s)
Scale-up and initiate validation of GMP manufacturing process at a scale compatible with USG requirements. Begin stability studies of the GMP product in formulation, dosage form, and container consistent with Target Product Profile. Initiate manufacturing process validation and consistency lot production. Conduct animal efficacy studies as appropriate. Conduct Phase 2 clinical trial(s).
7a. Conduct GLP animal efficacy studies as appropriate for the product at this stage.
7b. Complete expanded clinical safety trials as appropriate for the product (e.g., Phase 2).

- Submit study plans for CMC (chemistry, manufacturing, and controls) validation to FDA for review.
- Challenge material production, performance of assays for pivotal challenge efficacy studies may be performed in more than one facility as determined by specific product development considerations, resource availability, and FDA requirements.
- Study plans for large scale Phase 3 clinical trials are submitted to FDA (end of Phase 2 meeting).
- Submit plans (and proposed statistical analysis) for pivotal animal efficacy studies to FDA for review.
TRL 8: Completion of GMP Validation and Consistency Lot Manufacturing, Pivotal Animal Efficacy Studies or Clinical Trials, and FDA Approved or Licensure
Finalize GMP manufacturing process. Complete stability studies in support of label expiry dating. Complete pivotal animal efficacy studies or clinical trials (e.g., Phase 3), and/or expanded clinical safety trials as appropriate. Prepare and submit NDA/BLA. Finalize Target Product Profile in preparation for FDA approval.
8a. Complete pivotal GLP animal efficacy studies or pivotal clinical trials (e.g., Phase 3), and any additional expanded clinical safety trials as appropriate for the product.
8b. Prepare and submit New Drug Application (NDA) or Biologics Licensing Application (BLA) to the FDA.
8c. Obtain FDA approval or licensure.

TRL 9: Post-Licensure and Post-Approval Activities
9a. Commence post-licensure/post-approval and Phase 4 studies (post-marketing commitments), such as safety surveillance, studies to support use in special populations, and clinical trials to confirm safety and efficacy as feasible and appropriate.
9b. Maintain manufacturing capability as appropriate.

- Submit BLA/NDA to FDA for review. Submit plans for post-marketing studies (if applicable), any restrictions necessary to ensure safe use, patient information, labeling claims, product label, and promotional packages.
In future articles, we will discuss additional subcategories of biomedical TRLs (drugs, biologics, and vaccines; medical information management/information technology [IM/IT]). In addition, various TRLs are further defined for Biomedical and MCM applications, as well as TRLs developed or categorized by Federal Agency. All of the resources listed in this are also available at Tier Seven.
18 May
2018